
Millions of years of evolution have given us genomes that are like giant datasets for drug development. Finally, we are learning how to study them.
Clinical trials for new drugs are expensive and uncertain. More than nine out of every ten drugs that enter human trials fail because of harmful side effects or a lack of efficacy. Safety and effectiveness in animals often fail to translate to humans. Imperfect as animal trials are, though, we need to know what happens when we block or boost certain biological pathways with drugs.
But there is another source of this data that we are beginning to draw on. Nature has already done lots of the trials we want to run, through random genetic mutations and natural selection. Since the first full mapping of the human genome in 2003, and the rise of technologies to sequence human DNA, we now can study the genetic mutations in nature that do the same thing as the drugs we want to test. For the first time, we are starting to peer into nature’s laboratory and learn from the millions of years of experiments it has done.

Subscribe for $100 to receive six beautiful issues per year.
Trials and errors
On 13th March 2006, eight healthy young men began a clinical trial at Northwick Park Hospital in North London. A new drug called theralizumab was being tested on humans for the first time.
Theralizumab is a monoclonal antibody, a lab-made protein that mimics the body’s immune system antibodies. It is engineered to activate a type of immune cell in the blood called T cells. Normally, these cells are activated only when two proteins on their surface make a signal. These proteins are called the T-cell receptor and CD28. Theralizumab can circumvent this ‘two-factor authentication’ and activate the T cell directly through CD28 alone.
In early animal studies, the drug appeared to have many medical benefits, such as killing cancerous leukemia cells and dampening autoimmune responses in rheumatoid arthritis. Injecting the drug into crab-eating macaque monkeys, which are commonly used in drug testing because their physiology and immune systems closely resemble those of humans, suggested it could be safe even in higher doses than researchers planned to ever use. The UK’s Medicines and Healthcare products Regulatory Agency allowed TeGenero, the company developing the drug, to use it in human trials.
Volunteers were randomly assigned into either receiving theralizumab or a placebo, with infusions that were, relative to body size, 1/500th of the dose the macaques got. The volunteers received their infusions within minutes of one another. Within an hour, they all developed life-threatening reactions: their heads and necks swelled dramatically giving them an ‘elephant man’ appearance; they experienced severe pain, difficulty breathing, plummeting blood pressure, and racing heart rates. They ended up in the intensive care unit with multiorgan failure.
Further investigations found that the drug had activated T cells uncontrollably, flooding the volunteers’ bodies with inflammatory proteins called cytokines. There had been unseen subtle differences in the CD28 expression patterns between macaques and humans, which may have resulted in the monoclonal antibody triggering adverse reactions in humans but not in macaques. The resulting drop in investor confidence drove TeGenero to bankruptcy. The ‘elephant man’ trial stands out as a catastrophic failure in the history of clinical trials.
After the theralizumab tragedy, regulatory agencies implemented tighter clinical trial protocols. These included requiring sequential dosing, starting at much lower doses, with longer intervals between participants, and requiring more rigorous preclinical testing of biological drugs. Despite these stricter rules, serious adverse events continue to occur: between 2010 and 2015 UK clinical trials saw 7,187 adverse reactions, of which 493 were life threatening.
Nature’s laboratory
One option is to keep doing slower trials at greater cost. But nature often provides an alternative: random mutations produce the same changes researchers try to achieve with drugs, in effect providing millions of natural experiments.
The human genome is three billion base pairs long, and every individual inherits millions of genetic variants from their parents: places where they differ from the average person. They also get an additional 70 to 90 new mutations (on average), which they then pass onto their offspring.
With three billion base pairs and nearly eight billion humans alive today, any mutation that is compatible with life and reproduction likely exists in at least one person’s genome somewhere in the world. These naturally occurring mutations serve as a comprehensive catalog of genetic changes. The effects range from subtle changes in cellular processes to dramatic differences in enzyme levels, disease susceptibility, and other medically relevant traits.
Human genetic variations and the traits they cause (known as phenotypes) can be thought of as natural experiments. A genetic variant that inactivates a gene – a string of the genome that codes for a protein – can tell us about the potential effects of a drug that inhibits that same protein.
Take the example of DGAT1 inhibitors. The DGAT1 enzyme catalyzes the final step in synthesizing certain types of blood fat, and is essential for fat absorption in the intestine. Researchers hypothesized that inhibiting this enzyme would reduce fat absorption and promote weight loss. Preclinical studies in mice and rats supported this idea. However, during clinical trials with healthy human volunteers in 2011, many participants experienced severe diarrhea and vomiting, which led to the early termination of the trials.
As the DGAT1-inhibitor trials failed, pediatric doctors at Boston’s Mass General Hospital for Children found a clue as to why. They had been studying two siblings who had been born with a rare diarrheal disorder whose symptoms closely resembled those of the trial participants: healthy at birth, but developing severe diarrhea and vomiting on the third day after starting breast milk. Each child required months of intensive care and parenteral nutrition (tube feeding). Both became malnourished and suffered infections due to immunosuppression. The girl survived, but the boy died at eight months old. Genetic testing revealed both siblings carried two copies of a mutation that inactivated DGAT1. The researchers concluded that this lack of functioning DGAT1 had given their intestinal cells an extreme intolerance to food.
Genetic research can also identify new drug targets: proteins in the body that can be bound by drugs to treat diseases.
HIV enters human immune cells via the protein receptor CCR5. In 1996, researchers discovered that some people are immune to HIV because they have a genetic mutation that stops their immune cells from having this receptor. About 10–15 percent of Europeans have one gene with this mutation, and about two percent have two genes, which gives them immunity without making them unhealthy in any other way. This discovery led to the development of Maraviroc in 2007, which binds with people’s CCR5 receptors and stops HIV from entering their cells.
A 2015 study combined human genetic association databases with data on drug development programs and matched drugs with genes that coded for the same protein the drugs targeted.
The researchers tested how often a drug program that corresponded with real genes succeeded compared to those without. The odds of a drug getting regulatory approval doubled if there was supporting genetic evidence. A 2024 follow-up analysis found an even higher result: 2.6 times higher approval rates for genetically informed drugs.
Private investment in sequencing
Genotyping is the cheapest and most common method for mapping human genetic differences. But it only directly measures the 500,000 or so locations of the human genome where people most commonly differ, called ‘single nucleotide polymorphisms’ or SNPs (pronounced ‘snips’). Because the genetic variants that have significant disease relevance like the CCR5 mutation are rare, genotyping is not good at picking these up.
To uncover disease-relevant genetic variation, we needed to sequence large numbers of people and look at about tens of millions of locations. This means mapping the entire exome, which is the 1.5 to 2 percent of the genome that acts as instructions to cells about how to make proteins.
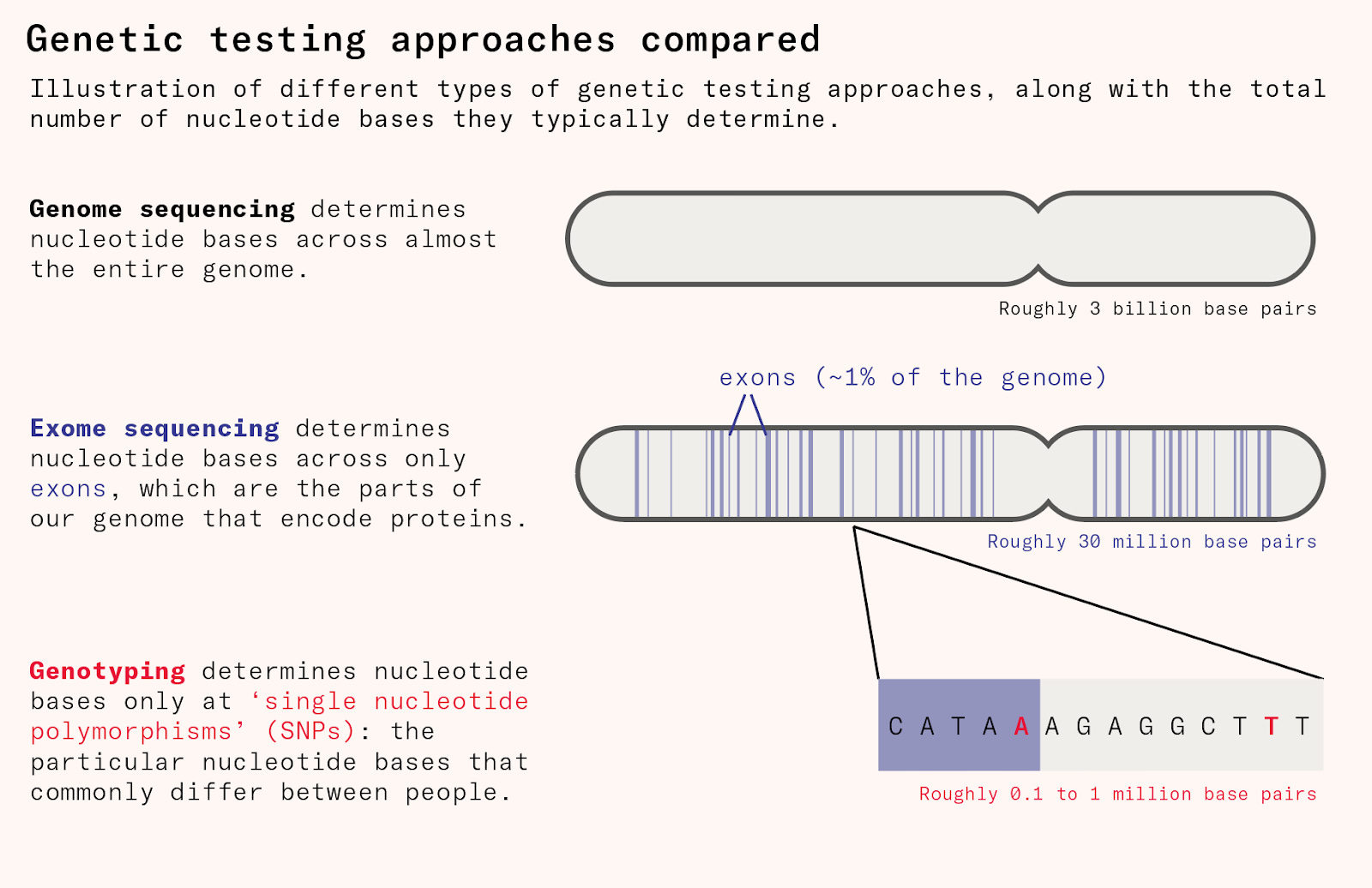
Only since the 2010s have drug companies begun to sequence people’s exomes at scale, allowing for much more efficient drug discovery. This began with projects such as DeCODE genetics, which includes genetic data on nearly half the population of Iceland, about 150,000 people, and links this information to disease diagnoses, blood tests, and various phenotypic data from nationwide registers. Before the rise of 23andMe and the UK Biobank, it was the world’s largest genetic database.
Using DeCODE, researchers discovered a mutation in the amyloid precursor protein that is common among Icelanders and which confers protection from Alzheimer’s disease. This discovery validated a drug class that was already under active development for the treatment of Alzheimer’s.
In 2013, the drug company Regeneron began using human genetics for drug discovery by sequencing the DNA of 100,000 people and linking this genetic data with medical records to identify genetic associations. Its scientists quickly uncovered a variant of a gene called HSD17B13 that conferred protection from liver disease. The gene encoded a liver-specific enzyme localized to fat droplets in the liver cells. Individuals carrying this variant, which gave them less of this enzyme, had a lower rate of chronic liver disease than average.
Pharmaceutical companies’ ambitions have continued to grow. In 2018, a group of pharmaceutical companies launched a project to sequence the exomes of half a million people. The participants came from the UK Biobank, a trove of longitudinal health information gathered since 2006, covering people who have already been genotyped. This 2018 population-scale exome sequencing has already proved enormously consequential, showing the protective links between variants in GPR75 with obesity, ANGPTL7 with glaucoma, CIDEB with liver disease, CHRNB2 with smoking addiction, and MAP3K15 with type 2 diabetes.
Diversity matters
Scaling up sample sizes and doing more thorough mapping are not the only ways to discover rare genetic variants in humans. There is a cost-effective alternative: diverse populations.
Human populations around the world differ in their genetic makeup thanks to geographical isolation and linguistic and cultural segregation. With rare exceptions, population groups were never completely isolated from each other, resulting in genetic diversity that exists mostly as a continuous spectrum rather than as distinct groups.
Genetic variants vary in their frequencies: what is common in one population group is rare in others. The CCR5 mutation is present in 10–15 percent of Europeans but less than two percent of Africans and South Asians have it. To discover its protective association with HIV infection, scientists would have needed to study significantly more people if they had studied only Africans and South Asians.
The same applies to variants that are common in other populations but rare in Europeans. The initial discovery of protective association between PCSK9 and low-density lipoprotein cholesterol was possible because the genetic variants were common in Africans but rare in Europeans. When researchers sampled 128 individuals from a multiethnic cohort with extremely low low-density lipoprotein cholesterol, more than half of the sample had African ancestry.
Early sequencing efforts focused predominantly on Europeans. Nearly 90 percent of the UK Biobank participants are of European ancestry, reflecting the UK’s historic ethnic makeup. Many sequencing projects have now started targeting exclusively non-European participants. Regeneron and AstraZeneca have recently partnered with academic researchers in the UK and Mexico to sequence 150,000 Mexicans. A group of drug companies has established Together for Change to build the world’s largest genetic database of people with African ancestry, with 500,000 African American participants. Similar initiatives are in the making around the world.
Human knockouts
These ethnically focused databases are already paying off. Since individuals carry their genetic variants throughout their lives, a mutation in humans can show us what the lifelong effects of taking a drug that had the same effects would be, giving us much better safety data than an early-phase drug trial. This is especially true when mutations affect both the functional copies of a gene, leading to a complete gene deficiency and making the affected individuals what are called ‘knockouts’.
Primary hyperoxaluria type 1 (PH1) is an extremely rare genetic disorder. People with PH1 lack the enzymes to detoxify glyoxylate, which oxidizes into a compound called oxalate. These oxalates cannot all be excreted through urine and form into kidney stones which steadily damage the kidneys. People with PH1 develop kidney failure early in their life and do not live beyond their twenties without dialysis or liver and kidney transplantation.
In the 2010s, Alnylam began developing a medicine for PH1. The idea was to block the activity of glycolate oxidase, the enzyme one step up in the chain, which turns glycolate into glyoxylate. Instead of glyoxylate, water-soluble glycolate would accumulate and be safely urinated out.
The approach worked in preclinical studies, and the researchers did not find any adverse effects. But researchers faced some important questions. Is the drug safe to give to humans to take over an entire lifetime? What are the long-term consequences of inhibiting glycolate oxidase? The answers came from a British Pakistani woman who enrolled in the Genes and Health cohort, a UK research project dedicated to individuals of South Asian origin. The researchers found that the woman was a knockout for the gene, HAO1, which encodes the glycolate oxidase enzyme. Her existence showed that a healthy life is possible without glycolate oxidase, and it is safe to switch off HAO1 permanently in humans. The drug passed through all phases of clinical trials and received Food and Drug Administration clearance in 2020.
The longstanding cultural practices of consanguinity (marrying with close relatives) and endogamy (marrying within a small community) make South Asians in India and Pakistan a particularly good source of these ‘human knockouts’ with no copies of a gene.
Researchers estimate that the odds of observing the same mutation that was found in the British Pakistani woman in an outbred population are 1 in 30 million. Many South Asians carry two copies of such mutations, and they can be found by sequencing only a few thousand individuals; to identify the same in an outbred population, one would need to sequence millions of individuals.
The Pakistani Genomics Resource is the largest South Asian cohort in the world, comprising more than 150,000 participants from communities known for their longstanding tradition of consanguineous marriages. Results from the first 10,000 participants, published in 2017, found at least one human knockout for 1,317 genes in the human genome, nearly twice the rate in the five-times-larger UK Biobank. A 2022 update found around 14,000 knockouts across more than 5,000 genes from looking at just 80,000 Pakistanis. To identify the same number of knockouts in an outbred population would have required more than 11 million individuals.
For many of those genes, dozens of knockouts have been identified, often clustering within a single extended family. In 2017 the Pakistani Genomics Resource reported a pedigree chart of a Pakistani family where 33 members were knockouts for APOC3, a major heart-disease-related drug target.
The family was Traced by Recontacting a participant from the database who was found to be an APOC3 knockout. Not only the participant, but his wife and all their nine children also lacked a functional copy of APOC3 – plus a further 23 members of their extended family. Not a single homozygous carrier for that same mutation has been found among the 500,000 UK Biobank participants.
One man’s curse, another man’s blessing
The focus on identifying beneficial mutations has led to a bias in genetic databases, favoring data from healthy people. This is useful for finding protective mutations. But by sequencing only healthy volunteers, researchers miss rare, disease-causing mutations, which can be just as helpful for drug development.
One example of such a mutation is the one that creates excessive fetal hemoglobin. Until birth, humans produce fetal hemoglobin, which binds aggressively to oxygen to ensure that a baby receives sufficient supply from its mother’s blood. After birth, humans start producing adult forms of hemoglobin, which bind less aggressively to oxygen. The switch from fetal to adult hemoglobin is controled by many genes, including BCL11A (discovered in 2007 through genetic association studies of fetal hemoglobin levels in blood).
Normally, adults stop making fetal hemoglobin and switch almost entirely to the adult type. But in some people, a little bit of the fetal version sticks around, helping to protect against sickle cell disease.
Vertex used this insight to design a gene-editing therapy: they edited blood stem cells to produce less BCL11A and so more fetal hemoglobin. The trick was figuring out how much to dial down BCL11A. Too much suppression kills blood cells, but too little doesn’t help.
The answer came from kids with a rare brain disease, who were born with only one working copy of BCL11A. Their blood naturally had more than enough fetal hemoglobin, well above the level sickle cell patients needed to fight their illness. This showed that cutting BCL11A by half in blood cells is sufficient to push the fetal hemoglobin levels above 20 percent. While partial BCL11A deficiency throughout the body from conception is harmful, selectively reducing BCL11A in blood cells is life-saving for sickle cell patients.
From exomes to genomes
Genotyping and sequencing have made it possible to find protective variants such as CCR5 and confirm drug safety thanks to healthy ‘human knockouts’ who live without key proteins. But the focus so far has been on protein-coding regions, which leaves much of the genome unexplored.
The future will involve sequencing not just the exome, the 1.5 to 2 percent of genes that code for proteins, but the entire genome. Once mistakenly deemed ‘junk DNA’, these non-coding regions affect how, when, and where coding genes work. Identifying non-coding mutations that disrupt a gene’s function in a specific tissue or at a particular developmental stage will help drug developers identify still more targets that are currently overlooked.
There are moves in this direction. UK Biobank completed whole genome sequencing of its entire 500,000 participants with funding from a consortium of drug companies. The National Institutes of Health’s All of Us Biobank opted for whole genome sequencing of its entire million participants and has completed sequencing of the first 250,000 individuals.
Natural experiments help scientists discover drugs and conduct safe trials. Our ability to study them depends on both what is sequenced and the amount of data from underrepresented human populations and patients with rare diseases.
By digging through new datasets, we will soon see whether non-coding genes tell us what the coding genome couldn’t. An era of rapid drug discovery awaits. By studying the genome we can use nature’s experiments to replace our own.