Antibody therapies are four of the world’s ten best selling drugs. If they were cheaper, they could prevent millions of deaths from rabies, malaria, and dengue.
Diphtheria, the strangling angel, was one of the great killers of children in the 19th century. The infection owes its lethality to a potent toxin released by the bacteria Corynebacterium diphtheriae. Diphtheria toxin destroys the heart, lungs, and liver. In the late stages of disease, the infected are suffocated by a buildup of dead, grey, tissue in their throats. Most victims were children under five; before modern treatments, as many as half of infected babies and toddlers died. There was little parents and doctors could do against the scourge, until biotechnology provided the first answer.
As the 19th century came to a close, Emil Behring, a Prussian doctor, took on the challenge of searching for a cure. Inspired by the nascent science of immunization and with no chemical treatment forthcoming, Behring looked to biology. In recent years, Louis Pasteur had demonstrated the power of immunization, with vaccines for chicken cholera, anthrax, and rabies. Somehow, exposure to an infectious agent (even in weakened form) transformed the blood of an exposed human or animal. Behring suspected this property could be transferable to other animals.

Subscribe for $100 to receive six beautiful issues per year.
Remove the red cells and clotting factors from any animal’s blood, and you are left with serum, a clear liquid roughly the color of apple juice. Behring suspected that serum carried the ‘antitoxins’ induced by vaccination. In 1890, Emil Behring and his Japanese collaborator Shibasaburo Kitasato showed that by injecting rabbits with diphtheria toxin, extracting their serum, and injecting it into a new rabbit, the receiving rabbits could survive an injection of a normally lethal dose of diphtheria toxin. Immunity had been transferred along with the serum.
{kind=link}
Describing his findings, Behring quotes the devil from Goethe’s Faust: ‘Blut ist ein ganz besonderer Saft’ (‘Blood is a juice of rarest sort’).
Human tests of Behring’s serum therapy followed shortly after: the treatment cured 77 percent of 220 children hospitalized with diphtheria. For this work, Behring became the recipient of the first-ever Nobel prize for physiology or medicine in 1901.
But serum’s promise still seemed limited. In his Nobel prize lecture, Behring expressed skepticism that the ‘disinfecting’ powers of serum therapy could ever be replicated artificially:
If now an internal disinfection has, nevertheless, been achieved, this is not thanks to speculation or change of doctrine, but thanks to the fact that Nature herself has been taken as a guide. I doubt whether it will ever be possible to establish artificially the antitoxic principle of serum therapy in diphtheria, without the aid of vital organization and secretion faculties.
Emil von Behring in his 1901 Nobel prize lecture
In the early days, serum was mass-produced in sheep, and then horses, which had greater blood volume (35–40 liters vs under five). Serum therapy was applied to many diseases, including pneumococcal bacterial infections (the most common cause of pneumonia), for which it was the only effective treatment before penicillin became widely available in 1945.

But serum therapy eventually fell out of favor. It was no longer worthwhile after the discovery of effective antibiotics in the late 1920s. Production was too variable; the batch release and manufacturing process were onerous; and allergic reactions (fevers, rashing, and swelling of joints) were frequent.
At the time of discovery and therapeutic application, Behring didn’t know which component in serum therapy was the active ‘disinfecting agent’. We now know that the antibodies were responsible. Antibodies are Y-shaped proteins tipped with exquisitely specific ‘lock and key’ targeting motifs that bind to foreign proteins in the blood or on the surface of cells. Antibodies neutralize the effects of their targets by binding and physically obstructing them, or by ‘tagging’ the target for removal by immune cells.
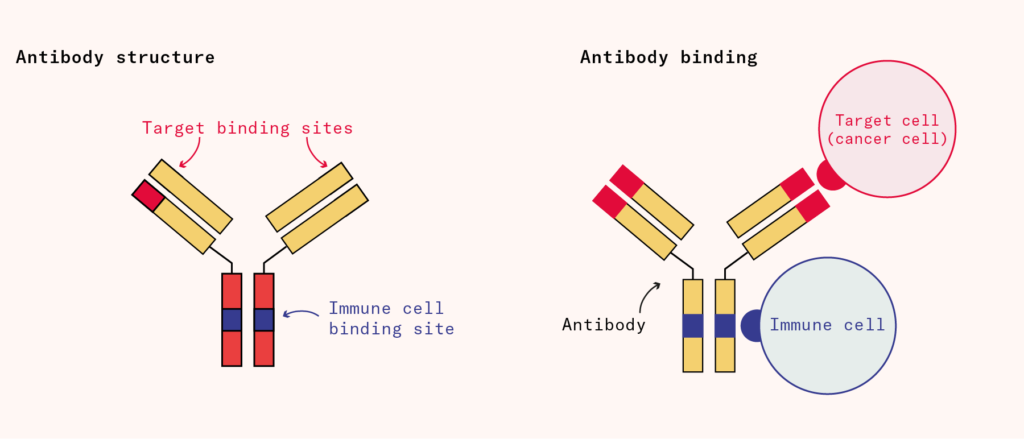
Antibodies have many properties that make them attractive therapeutics, among them that they can circulate for weeks or even months in the bloodstream, hunting for their targets. By selecting randomly from a repertoire of genetic fragments, immune cells can find suitable shapes for the ‘variable’ region of the antibody to make it bind to almost any invading protein with exquisite precision and specificity. Serum contained a mixture of useful antibodies along with many others, so large quantities of serum were needed to ensure sufficient amounts of therapeutically relevant antibodies were present.
The potential of antibody therapy was there, but predictable chemistry (in the form of antibiotics) had rendered the messy biological process of serum obsolete. To make antibody therapy useful again, the active component would need to be isolated, purified, and controlled.
Today, Behring has been proven wrong: antibody therapeutics are a mainstay of treatment for many diseases. The highest revenue drug in the world is Keytruda ($30 billion in sales in 2024), a monoclonal antibody used to treat almost every type of solid tumor. Merck & Co (Keytruda’s manufacturer) estimates that 2.5 million people have been treated with the drug as of 2024. In all, four of the ten best-selling drugs are monoclonal antibodies.
Modern pharmaceutical forms are no longer limited to synthetic chemicals dispensed in neat pill boxes. The bulky ‘biologic’ class of drug molecules (large, complex therapeutics produced in living organisms e.g. proteins, whole cells, and tissues), which includes antibodies, has come to the fore.
But it was just in the past decade or so that the technology matured enough to give us multiple new approvals per year; the FDA only recently approved the 100th monoclonal antibody drug. To turn antibodies into a scalable technology, as we had done previously with small synthetic molecules, we needed to stack many improvements in manufacturing. With these advances, came synthetic processes that made far more diseases treatable.
Making monoclonals
To industrialize serum production, it was first necessary to identify the active component in serum therapy.
The journey started with the microbiologist Rudolf Kraus’s observation in 1897 that the introduction of a foreign protein into the blood would lead to the formation of a cloudy precipitate. This became known as the ‘precipitin’ reaction.
In the 1920s, the organic chemist Michael Heidelberger and physician Oswald Avery isolated the substance from the precipitin reaction, and determined that they were proteins based on their nitrogen content. ‘It appeared to me that there was a crying need to determine the true nature of antibodies’, Heidelberger wrote in a memoir, ‘and that until this was done there could be no end to the polemics and uncertainties that were plaguing immunology’.
Some time later, in 1945, the immunologist Thomas Harris and colleagues at the Philadelphia medical school found that antibodies were produced by a type of white immune cell in the blood. With the nature and site of production of antibodies identified, scientists had begun to make real progress on understanding the mysterious substance responsible for the activity of Behring’s serum therapy.
Specifically, it turned out that plasma cells (a type of white blood cell) secreted antibodies into the blood. In myeloma, a form of blood cancer, a single malignant plasma cell divides uncontrollably. These cells produce many copies of a single antibody or antibody fragment (the ‘m-protein’). The overproliferation of malignant plasma cells leads to large quantities of m-protein in the blood of patients and overwhelms the diverse ensemble of white blood cells that would otherwise be present in the blood.
Because myeloma cells spew off large quantities of antibodies as they grow, they were seen as a useful tool to study antibodies. However, while it was easy to obtain some quantity of antibodies from these cancerous cells, there wasn’t an easy way to get many copies of any particular one.
Even if the exact design for an antibody had been known, which it wasn’t, the sort of chemical synthesis we might do to produce a ‘small molecule’ drug like aspirin wasn’t feasible. As ‘biologics’, antibodies are thousands of times bigger than a drug like aspirin, and have much more complex structures. Even today, our chemical techniques lack the fine-grained control necessary to assemble all the pieces in the right order at scale. Entering the mid-20th century, the only viable path forward was to hijack existing cellular machinery and redirect it to our own ends.

Limited progress was made until the 1970s, when the immunologist Georges Köhler, and biochemist César Milstein at the Laboratory of Molecular Biology (known as the LMB) wondered if they could solve the problem by taking normal plasma cells that produced a specific antibody and fusing them with myeloma cells to create immortal cells that produce many copies of a desired antibody. With the help of their technician Shirley Howes, Köhler and Milstein managed to create these fused cells, which they called ‘hybridomas’.
Each hybridoma constantly produces a large amount of one specific antibody, so their products are called monoclonal antibodies (mAbs for short), meaning they come from a single clone or source. Although they were originally conceived as a research tool, hybridomas made it possible in principle to scale up antibody production for large-scale medical use, as long as you could identify and isolate which plasma cell produced a therapeutically relevant antibody.
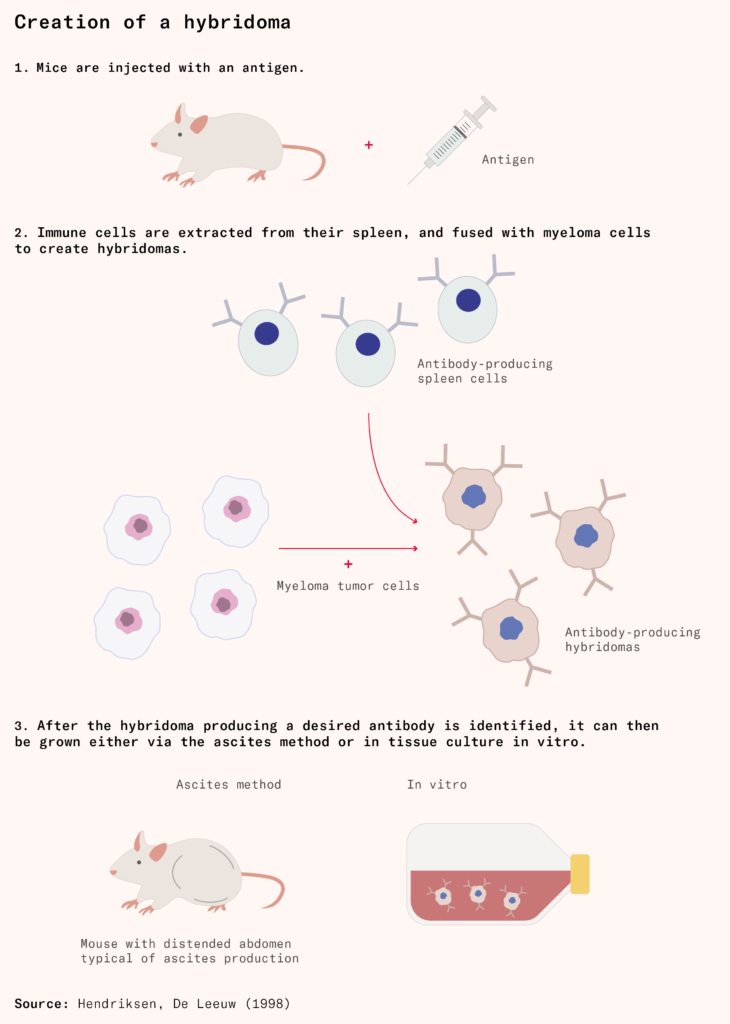
Initially, the most reliable method for growing hybridomas was the in vivo ascites method. In the ascites method, technicians or scientists injected hybridomas into the abdomens of mice, where they multiplied, bloating the mice with fluid and antibody-producing cells. There is usually a small amount of fluid in the abdominal space: less than 50 milliliters in humans, and 0.05 milliliters in mice, about the volume of a raindrop. But in the ascites method, mice can accumulate as much as ten milliliters of fluid in their abdomen, about a fifth of their body weight.
After growth, technicians or scientists repeatedly extracted fluid containing the antibody from the abdomen of the mice with a needle until it filled up again. The ascites method was cruel and painful for the mouse, which would eventually die of the induced tumor. But it was effective at producing highly concentrated monoclonal antibodies.
It was in these mouse micro-factories that Ortho Pharmaceuticals produced the first therapeutic monoclonal antibody approved for human use: muromonab, approved by the FDA in 1986 to prevent the rejection of kidney transplants.
The first FDA-approved monoclonal antibody
Ortho was best known for marketing a range of pregnancy tests. Ortho scientists hadn’t originally set out to create a monoclonal antibody therapy. Rather, muromonab was one of a series of antibodies intended as research tools to probe and better understand immune cell diversity and function.
Muromonab was found to block a key structure on T-cells, a protein called CD3. Blocking this protein inhibited the cells’ function, depleting the number of functional circulating T-cells. Ortho realized that these T-cells were responsible for organ rejection, and clinical trials of muromonab demonstrated that it could prevent organ rejection in transplant patients in a more targeted fashion than broad-spectrum immunosuppressants.
Muromonab’s clinical trials helped show that monoclonal antibodies could be useful drugs for conditions beyond infectious diseases. But the trials also helped to identify two major obstacles for the drug class as a whole.
Mouse-derived monoclonal antibodies would often be rejected by the human immune system. Antibodies produced by mice and by humans are subtly different, and this difference can be detected by the human immune system. Mouse-derived monoclonal antibodies like muromonab could only be dosed a few times before our immune system would develop its own neutralizing antibodies against the foreign monoclonal ones. In muromonab’s early trials, the drug staved rejection off for a week or two before the body rejected the foreign monoclonal antibody as well, after which traditional immunosuppression was required. Monoclonal antibodies from mice also circulate for just a few days in our blood, compared to the weeks or months for human antibodies. This meant that unreasonably high chronic doses would be needed for therapeutic effect.
Although the mouse manufacturing method worked for the low quantities needed for research and muromonab, it was not viable as a long-term solution. It was both too impractical and too cruel: scaling would have required tiling the ground with giant breeder factories of engorged mice, and few biotechs were keen to follow in Ortho’s footsteps.
It would take another 10 years for the next full-length monoclonal antibody to be approved by the FDA when, on November 26, 1997, rituximab was developed by IDEC Pharmaceutical Corporation and Genentech. The first step was antibody ‘humanization’. Researchers had attempted to work with human hybridomas, but the cells were unstable and the prospect of injecting humans with antigens and then harvesting their spleens was unlikely to get past the ethics committees. Instead, a molecular biologist called Gregory Winter pioneered a technique to ‘swap out’ the parts of antibodies responsible for binding from mice antibodies onto a human scaffold, preventing immune rejection.
Chinese hamster ovaries
Scaling up the manufacturing of monoclonal antibodies beyond the mouse method required the development of an entirely new manufacturing methodology. In the 1980s, scientists at Genentech were able to grow peptide and protein products like insulin in E. coli bacterial cells. Genentech’s vision was to make biologic production as straightforward as chemical synthesis.
Genentech led the development of recombinant DNA technology, which involves inserting genes from one organism into another to produce desired proteins. Scientists could take the human gene for insulin, insert it into bacterial DNA, and then grow the bacteria in large vats. The bacteria, now containing the insulin gene, would produce human insulin as though it were their own protein. This breakthrough allowed the mass production of human proteins without needing human or animal tissue sources.
But it soon became clear that bacteria could not produce every medically relevant protein. Genentech tried to use the technology to produce tissue plasminogen activator (tPA), a natural protein that dissolves blood clots and is used in medicine to restore blood flow following a stroke. Having a reliable supply of tPA – ‘clot buster’ – meant that emergency rooms could quickly administer the protein to restore blood flow, avoiding the brain tissue death that sets in within minutes, potentially preventing permanent disability or death.
But the attempts to try and manufacture tissue plasminogen activator in E. coli cells were a failure. The tPA protein is roughly 12 times larger than insulin. It didn’t fold correctly in bacterial cells and was too big to escape from bacterial membranes. This meant that breaking open the bacterial cells and purifying the protein from the other gunk required expensive extraction and post-processing steps.
This problem wasn’t limited to tissue plasminogen activator: proteins are not processed in the same way in bacterial cells as they are in human cells. Proteins need to fold into the right conformation to perform their functions, but this process doesn’t work all that well in bacteria. Many mammalian proteins, for example, are ‘sugar coated’: In antibodies, this affects how they bind to immune cells. But bacteria lack the machinery necessary to attach these sugars to proteins.
Complex proteins like tissue plasminogen activator have to be manufactured in mammalian cells. The solution, once again, came from rodents. In the early 1980s, a cohort of researchers across Stanford, Columbia, and MIT discovered how to amplify genes in the extracted ovary cells of Chinese hamsters, boosting the yields of recombinant proteins.
Along with humanization and recombinant DNA, the Chinese Hamster Ovary (known as CHO) became the key technology for biologic manufacturing at scale.
An optimized CHO cell line could secrete twice as many antibodies per cell per day than hybridomas, and sometimes as many as ten times more. Despite the high yields, there still wasn’t an easy way to grow them at scale. Genentech assumed that mammalian cells couldn’t grow well in the large steel fermentation tanks that yeast or E. coli grew in. Mammalian cells aren’t as hardy as E coli or yeast cells, which have cell walls, so researchers thought the cells would inevitably break during the stirring process which distributes nutrients and oxygen across the cells. Instead, mammalian cells were initially grown in smaller ‘roller bottles’, which gently aerated the cells as they turned over.

A more scalable solution was needed. A chemical engineer suggested growing the Chinese hamster ovary cells in a fermenter tank, while spinning the impeller ‘very, very slowly’ to prevent the movement from splitting the fragile cells. Chinese hamster ovary cells grow more slowly than E coli, so they didn’t need to receive as much oxygen. Surprisingly, it worked, and Genentech was able to produce enough tPA for their trials and, subsequently, their commercial launch. On November 13, 1987, Genentech’s tissue plasminogen activator became the first FDA-approved pharmaceutical product manufactured in Chinese hamster ovary cells.
Many companies followed where Genentech led, to total Chinese hamster ovary cell dominance: 70–80 percent of biologics are manufactured in these cells today.
Vitamin R
Using mammalian cells as production systems unlocked the ability to manufacture a whole new class of proteins at industrial scale, including antibodies. Recombinant technology and hamster cells had done for biologics what the German chemical factories had done for dyes: render a messy biological process into a scalable industrial one.
Genentech now needed good ideas for what to manufacture; Californian biotech company IDEC supplied one. IDEC had been working on what would become rituximab, spurred by the discovery that cancerous blood plasma cells could be targeted by antibodies. IDEC had manufactured small batches of rituximab and ran a few early clinical trials, and the early results were promising: Some patients with follicular lymphoma saw their tumors shrink by more than half after a single dose.
But IDEC was having trouble taking rituximab to market. Back then, investors were not enthusiastic about cancer drugs, suspecting both that the market would be too small and that IDEC lacked the capacity to scale up manufacturing and commercialize the drug. ‘All the analyses showed that this was a ridiculously small market’, recalled David Maloney, one of the doctors involved in early rituximab trials. ‘People could not imagine that this would be a marketable drug. They’d been turned down by everybody’.
Given their expertise with CHO cells, Genentech seemed a natural partner. Although nobody had manufactured a drug as complex as rituximab, Genentech had the most experience with the nascent technology. IDEC proposed a partnership in which Genentech would contribute funding and manufacturing expertise in exchange for a share of revenues. After looking at the trial data, Genentech was sold.
The initial rituximab manufacturing process was not suitable for the volume and predictability required to sell the drug at scale. The teams at IDEC reconfigured their process to use Chinese hamster cell lines instead of hybridomas. They also replaced the medium rituximab was cultured in, replacing fetal bovine serum (cow fetus fluid), a byproduct of the meat industry that contains substances that encourage cell growth, with an entirely synthetic growth medium. Rituximab was approved in 1997. The final dose, three whole grams of recombinant proteins, would have been unthinkable without the scaling improvements between hybridomas and Chinese hamster ovary cell lines.
Subsequent improvements in mammalian cell manufacturing have driven the cost of manufacturing recombinant monoclonal antibodies down by further orders of magnitude: today, monoclonal antibodies sell for between $50 and $150 per gram at commercial scale (a typical dose of a monoclonal antibody is a few hundred milligrams).
Rituximab’s impact on medicine extends far beyond its initial use in follicular lymphoma. The drug is a cornerstone therapy for numerous B-cell lymphomas (white blood cell cancers), transforming previously fatal cancers into manageable conditions, and features in so many regimens that specialists sardonically began calling it vitamin R. It has even found use in autoimmune conditions like multiple sclerosis, rheumatoid arthritis, and lupus.
Monoclonal antibodies for everyone else
Contrary to Behring’s expectations, it has turned out that there is no particular magic about blood or ‘vital organization’. It’s likely that we will continue to replace messy biological production with engineered systems, and drive down costs, increase scale, and expand the application of mAbs.
Therapeutic antibodies may be a 100-plus-year-old technology; but they are only now exiting infancy. They have much further to go.
For one, increased yields have shifted the bottleneck to the processing and purification of the final product. Most companies have a standard process that involves capturing the mAb, filtering out impurities, and inactivating any viruses. A particularly expensive material in this process is the filter meant to remove parvovirus, an extremely small DNA virus that infects Chinese hamster cells. The specialized nanofilters required to capture these particles are used once, thrown away, and add two to three dollars per gram to production costs. There has never been a documented case of parvovirus transmission from manufactured antibodies to humans, but regulators require the filter because of the hypothetical risk to immunocompromised patients.
There is probably more low-hanging fruit like this. There has been little pressure on companies to aggressively constrain the cost of goods. High drug prices in developed economies mean that monoclonal antibody gross margins can be as high as 90 percent, and the price of antibody therapies is practically uncorrelated from the cost of goods.
Some production systems have already achieved costs as low as $30 per gram, and continuous rather than batch manufacturing can shave another 50 percent off the costs of manufacturing. The Gates Foundation thinks even bigger: it recently put out a ‘grand challenge’ to solicit proposals for technologies with the potential to reduce the cost of monoclonal antibody manufacturing to $10 per gram.
Antibody design today relies on animals to produce antibody genes, which we put into a production system like CHO cells. The David Baker lab at the University of Washington has demonstrated that artificial intelligence can now design antibodies from scratch, eliminating the need for animal immunization or screening libraries. While traditional antibody discovery might screen millions of candidates to find one that works, computational design can predict binding virtually and generate optimized antibody sequences, which ideally will require lower doses.
Another way to reduce costs is by making antibodies smaller. Nanobodies, which are antibody fragments that contain just the binding region, are about one-tenth the size of full-length antibodies but retain specificity and the ability to bind and neutralize their targets. These mini-antibodies already naturally occur in camels, llamas, and alpacas.
Under $10 per gram, it becomes viable to develop monoclonal antibodies for new disease areas at a global scale. There are many diseases for which monoclonal antibodies could save millions of lives, including malaria, rabies, dengue, and respiratory infections. Monoclonal antibodies are particularly effective at treating infectious diseases. They are also an important part of pandemic preparedness: Donald Trump, for example, was treated with Regeneron’s anti-COVID monoclonal antibodies when he caught the virus.
Another target for cheap antibodies will be snakebites, which kill around 100,000 people per year. Antivenom that targets venom from multiple species is expensive and hard to reach for the people who are most affected, typically poor farmers in rural areas. Cheaper and more temperature-stable monoclonal antibodies can be stored and administered in the field.
A world in which monoclonal antibodies are distributed as ubiquitously and cheaply as aspirin could transform how we deal with almost every sickness: infectious disease and future pandemics, as well as autoimmune disease and cancer. Making this a reality requires costs to decrease by another order of magnitude (to well below $10 per gram). Separately, manufacturing capacity would need to increase from 30 metric tons per year today to hundreds or thousands of tons. While such a world was far beyond Behring’s imaginings, it is now within sight.